黑龙江大学井立强团队AEM:具有Co-N4位点的超薄酞菁钴/聚(庚嗪酰亚胺)异质结高效光催化二氧化碳还原和电子动力学研究
第一作者:王国薇,张洪光
通讯作者:李卓,张紫晴,井立强
通讯单位:黑龙江大学
论文DOI:10.1002/aenm.202405622
全文速览
聚(庚嗪酰亚胺)(PHI)作为一种新兴的g-C3N4(CN)替代材料,在光催化还原CO2领域展现出巨大潜力,但其电荷分离能力弱和电子诱导还原反应效率低的问题限制了其实际应用。为解决这一问题,本文以CN为前驱体,通过熔盐法可控合成了超薄PHI纳米片,并进一步利用π-π相互作用将酞菁钴(CoPc)聚集体组装到PHI表面,构建了CoPc/PHI异质结。优化后的CoPc/PHI异质结表现出优异的光催化性能,其CO生成速率达到116 μmol g-¹ h-¹,选择性高达97%,与CN和PHI相比,活性分别提升了23倍和15倍。实验与理论计算结果表明,光生电子从PHI向CoPc配体转移,促进了电荷的高效分离,随后电子进一步迁移至Co-N4活性位点,驱动CO2的高效转化。此外,*COOH中间体的低形成能垒和CO的快速脱附过程共同促进了CO的高选择性生成。原位瞬态吸收光谱分析表明,CoPc/PHI在CO2还原反应中的电子转移效率达到39.7%,远高于PHI的17.7%,这凸显了CoPc聚集体作为高能电子接受平台和催化活性位点的双重作用。本研究为设计高效异质结光催化剂以实现太阳能燃料生产提供了一种可行的策略。
背景介绍
化石燃料的大量开采,致使二氧化碳大量排放,加剧了温室效应,破坏了碳循环,这引起了人们的极大关注。利用太阳能将二氧化碳转化为高附加值化学品和燃料,是解决这些问题的一项前景广阔的技术。合理设计和开发有效的光催化剂,是实现高活性和高选择性二氧化碳转化的关键。PHI 正逐渐成为传统 CN 的替代品,有望实现高效的二氧化碳转化。但它的光催化性能仍受限于电荷分离能力弱和活性位点稀缺。构建异质结可有效促进 PHI 的电荷分离。过渡金属酞菁 MPcs 因能级合适,且拥有 M-N4 类单原子位点,成为理想的候选半导体。其中,CoPc 的 * COOH 形成能垒较低,*CO 结合能适度,有利于实现 CO2到 CO 的选择性转化。因此,构建维度匹配、界面连接紧密的超薄 CoPc/PHI 异质结,有望实现高活性和高选择性的 CO2光转化。此外,利用原位时间分辨技术揭示电荷转移和催化机理意义重大,但极具挑战。
本文亮点
1)通过π-π相互作用和静电组装将CoPc聚集体负载到PHI上用于无需任何牺牲剂的纯水体系CO2光还原。
2)优化后的CoPc/PHI异质结实现了116 μmol g-1 h-1的CO生成速率和97%的选择性,与原始CN和PHI相比,活性分别提高了约23倍和15倍。
3)原位μs-TAS深入揭示了CoPc/PHI电子转移效率的提高(从PHI的17.7%提高到39.7%)和电子寿命的延长对二氧化碳还原的贡献。
4)CoPc作为高能电子接受平台以及Co-N4位点的选择性CO转化能力,助力了高活性和高选择性CO2光还原。
图文解析
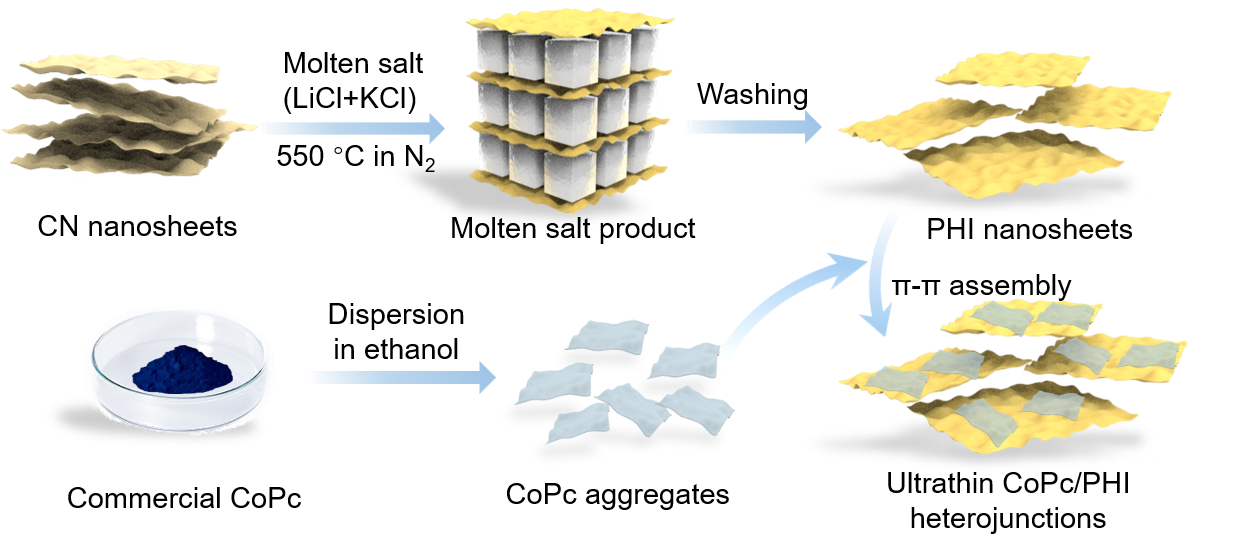
Scheme 1. Schematic illustration of the synthesis process for CoPc/PHI heterojunctions.
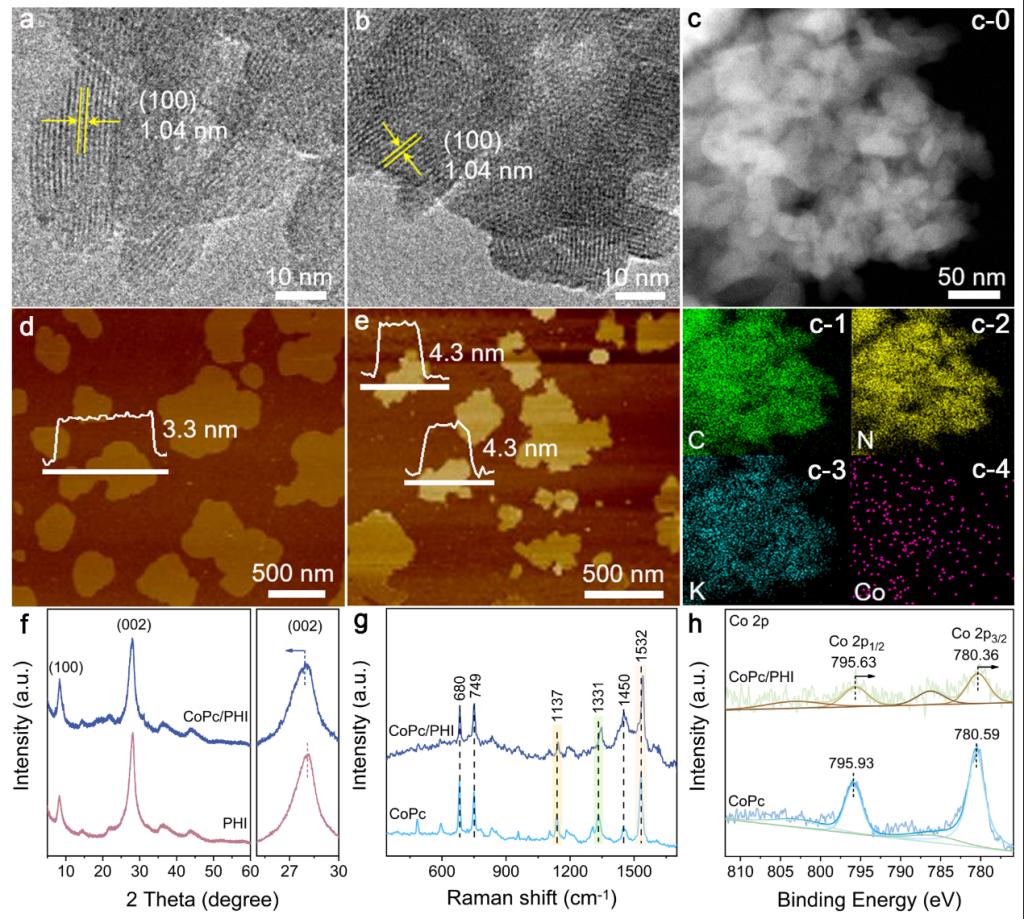
Figure 1. HR-TEM images of (a) PHI and (b) CoPc/PHI, (c) TEM image of CoPc/PHI and the corresponding EDX mapping images of elemental C, N, K and Co, AFM images and the corresponding height profiles of (d) PHI and (e) CoPc/PHI, (f) XRD patterns of PHI and CoPc/PHI, (g) Raman spectra and (h) XPS analyses for Co 2p of CoPc and CoPc/PHI.
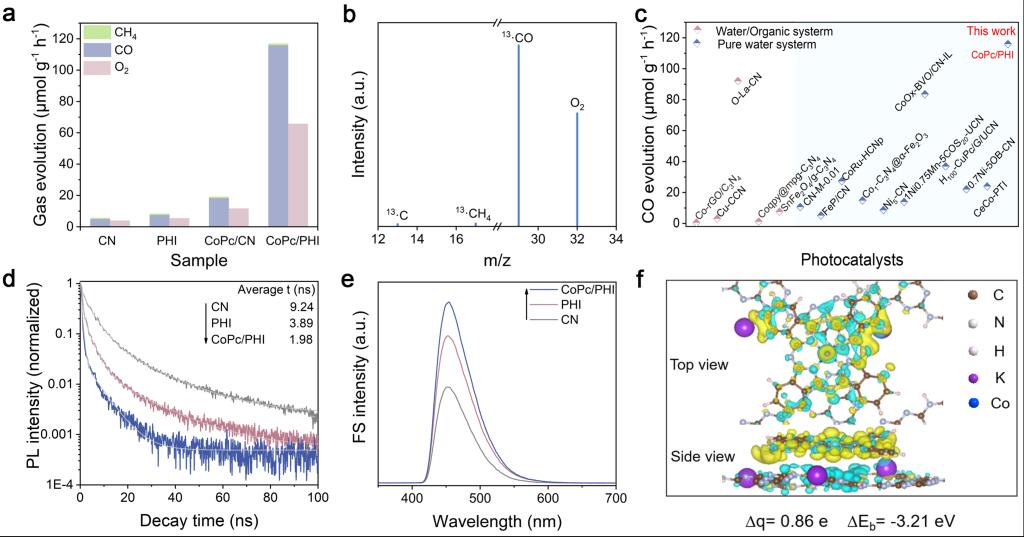
Figure 2. (a) Photocatalytic activities for CO2 reduction of CN, PHI, CoPc/CN and CoPc/PHI, (b) mass spectra of the products from the photocatalytic reduction of 13CO2 over CoPc/PHI, (c) comparison of CO evolution rates for recent reported photocatalysts and CoPc/PHI, (d) TR-PL spectra and (e) FS related to the produced hydroxyl radicals of CN, PHI and CoPc/PHI, respectively, (f) charge density difference plot of CoPc/PHI (The blue and yellow colors indicate charge depletion and accumulation, respectively, the isosurface value is 0.0005 e Å-3).
通过π-π相互作用,将CoPc聚集体可控组装于 PHI 纳米片上,成功构建出具备紧密界面连接的超薄 CoPc/PHI 异质结。CoPc在PHI表面的厚度约为2-3层,二者凭借π-π相互作用形成紧密连接的界面。优化后的CoPc/PHI异质结实现了116 μmol g-1 h-1的CO生成速率,选择性高达 97%。相较于 CN 和 PHI,其光活性分别提升了23倍和15倍。13C标记同位素实验证实,CO源于光催化作用下的CO2还原,而非CoPc或PHI的分解。CoPc/PHI 的 CO 生产速率优于纯水和有机体系中已报道的 CN 基和结晶 CN 基光催化剂。TR-PL和FS测试表明,CoPc/PHI 具有优异的电荷分离性质。理论计算显示,PHI 扩展的π共轭结构和离域的π电子增强了与CoPc间的π-π相互作用。
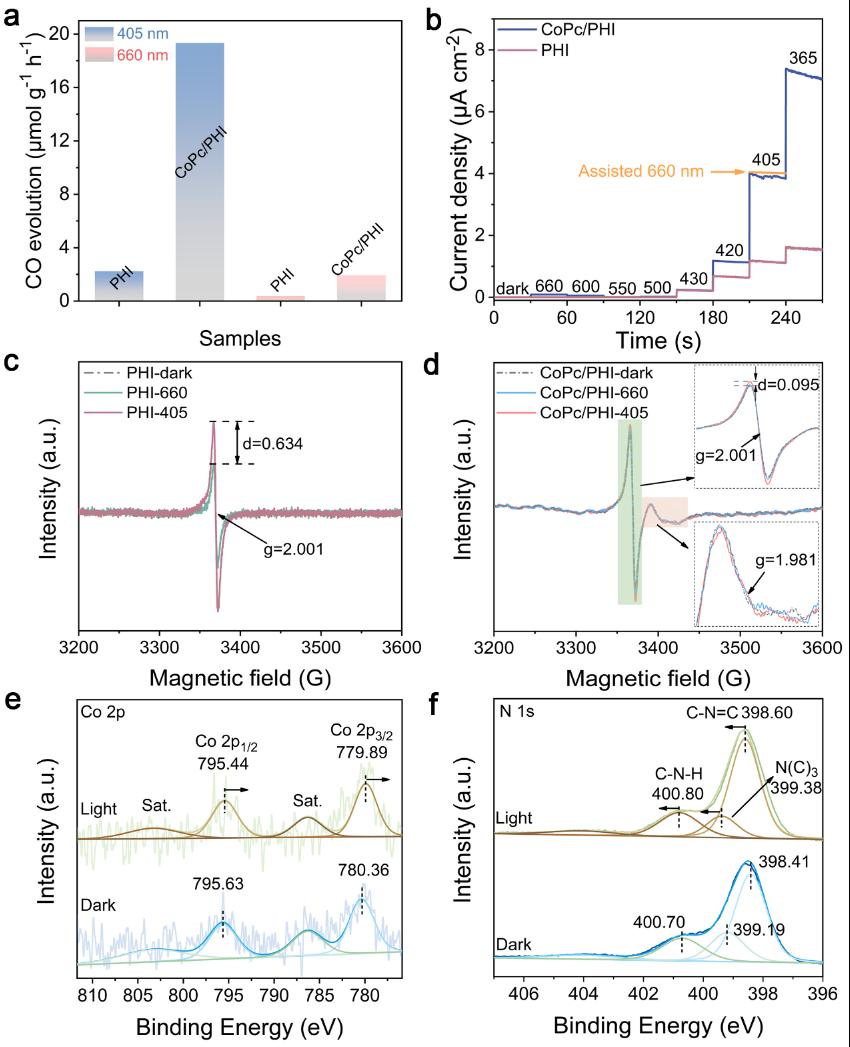
Figure 3. (a) Photocatalytic activities for CO2 reduction to CO under the irradiation of 405 and 660 nm light for PHI and CoPc/PHI, respectively, the light intensities were normalized the same, (b) the monochromatic photocurrent action spectra of PHI and CoPc/PHI, EPR spectra of (c) PHI and (d) CoPc/PHI under dark and light irradiation of 405 and 660 nm, respectively, in situ XPS spectra of (e) Co 2p and (f) N 1s of CoPc/PHI before and after 405 nm light irradiation.
在单波长光照条件(405和660 nm)下的 CO2 还原活性测试,揭示了PHI和CoPc之间可能存在从PHI到CoPc的高能平台电荷转移现象。单色光电流作用光谱进一步揭示了异质结内部的电荷转移机制。当激发波长大于500 nm时,PHI无法被激发,仅显示出微弱的光电流信号。当PHI和CoPc/PHI被430 nm光激发时,二者呈现出近乎相同的光电流密度,这意味着异质结内部尚未发生电荷转移。值得关注的是,与PHI相比,CoPc/PHI异质结在420 nm光激发下,光电流密度急剧上升。从其能带结构来看,该波长光照提供了充足的热力学能量,促使光生电子从PHI转移至CoPc。随着激发波长从420 nm进一步减小到405 nm,光电流密度大幅增加;在365 nm光照下,光电流密度更高,这表明电荷分离得到了进一步改善。借助低温电子顺磁共振(EPR)技术,对异质结体系中的电荷转移机制展开了探究。EPR 结果为PHI到CoPc的高能平台电荷转移机制提供了有力支撑。原位光照X射线光电子能谱测试进一步证实,PHI的光生电子向CoPc发生了转移。光照条件下,Co 2p 向低结合能方向偏移,这表明中心金属 Co 在光催化反应中充当电子受体,进一步印证了光电子先从PHI转移到CoPc配体,随后再转移到中心金属Co。
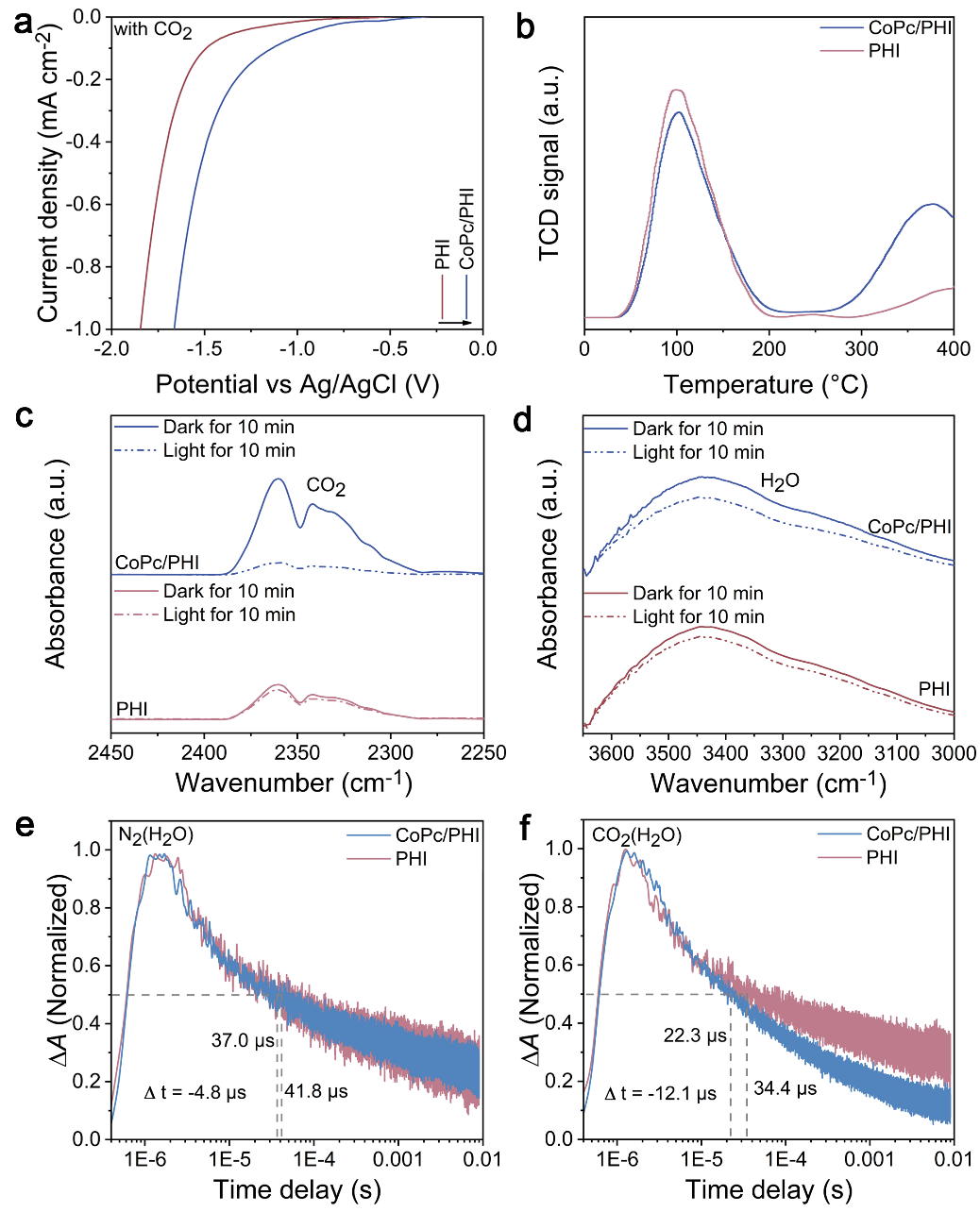
Figure 4. (a) Electrochemical reduction curves in CO2-bubbled systems, (b) CO2-TPD curves, in situ DRIFTS for (c) adsorbed CO2 and (d) H2O in dark for 10 min and under light irradiation for 10 min, µs-TAS decay kinetics in (e) N2(H2O) and (f) CO2(H2O) conditions for PHI and CoPc/PHI, respectively.
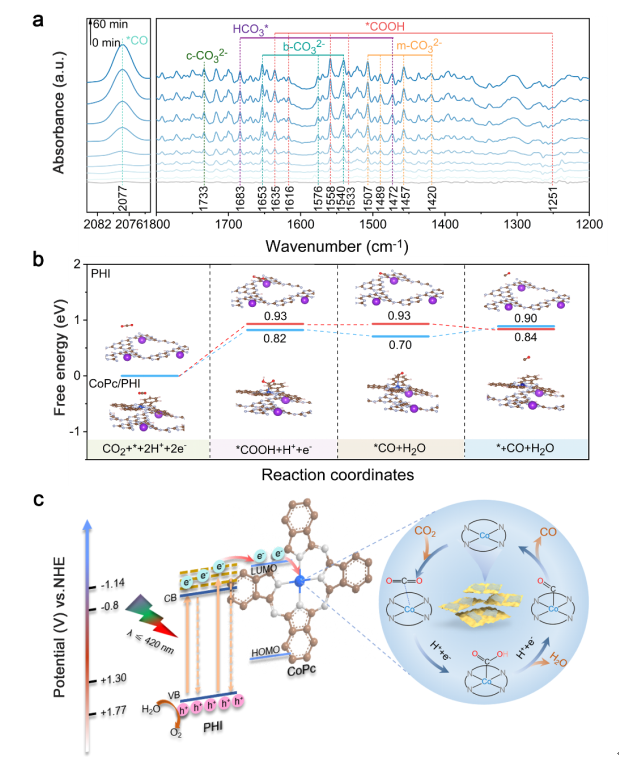
Figure 5. (a) In situ DRIFTS for the detected intermediates of CoPc/PHI under light irradiation, (b) Gibbs free energy diagrams and reduction pathway of CO2 to CO over PHI and CoPc/PHI, (c) schematic of transfer and separation of photogenerated charge carriers in the fabricated CoPc/PHI heterojunctions and the corresponding pathway for CO2 reduction reaction. (Brown, gray, pink, red, purple, and blue balls represent C, N, H, O, K and Co atoms, respectively)
实验结果显示,CoPc的引入有助于CO2的吸附与转化,而水的吸附和活化主要在PHI上进行。原位μs-TAS明晰了光催化CO2还原过程中的电荷转移机制和动力学过程。经计算,CoPc/PHI在CO2还原反应中的电子转移效率可达39.7%,远高于PHI的17.7%。随后,利用原位红外技术进一步探究光催化CO2RR过程中涉及的中间活性物种变化。光照过程中,*COOH 和*CO吸收峰逐渐出现且增强,这意味着CO2发生了有效转化。DFT计算表明,CoPc的引入可显著降低 *COOH 的形成能垒,同时,Co-N4位点处较低的*CO结合能,推动了 CO2向CO的选择性转化。基于上述结果,绘制了 CoPc/PHI 异质结电荷转移及其催化转化CO2过程的机制图。在波长 ≤ 420 nm 的光激发下,PHI 被激发产生高能量激发电子(HLEEs),这些电子随后转移至 CoPc 的配体上,再转移到Co-N4位点,通过形成*COOH实现CO2转化,最终生成CO,而位于PHI脱质子化酰亚胺桥附近的空穴则会通过质子耦合电子转移过程氧化 H2O 产生 O2。
总结与展望
本文依托熔盐法可控合成了PHI纳米片,借助π-π相互作用和静电组装,将CoPc聚集体负载于PHI之上,构建了维度匹配且界面连接紧密的CoPc/PHI异质结,用于高活性、高选择性的CO2光还原。优化后的异质结CO生成速率达116 μmol g-1 h-1,选择性为97%,相较于CN和PHI,光催化活性分别提升约23倍和15倍。性能提升主要源于光生电子从PHI到CoPc配体,再到Co-N4 (II) 位点的高效电荷转移与分离,以及Co-N4 (II)处将CO2高活性、高选择性地转化为 CO。经原位μs-TAS计算,CoPc/PHI二氧化碳还原的电子转移效率(ETE)为 39.7%,远高于PHI的17.7%,凸显了CoPc聚集体作为电子接受平台和催化位点的双重功能。这项工作为设计基于PHI的异质结提供了可行途径,能高效地将CO2转化为有价值的产物CO,而CO作为后续还原碳氢化合物的关键中间产物,为各类燃料生产奠定了基础。
文献信息
Ultrathin Cobalt Phthalocyanine/Poly(heptazine imide) Heterojunctions with Co-N4 Sites for Improved Photocatalytic CO2 Reduction and Electron Kinetics. Guowei Wang#, Hongguang Zhang#, Tianyu Tang, Zhijun Li, Yang Qu, Zhuo Li*, Ziqing Zhang*, Liqiang Jing*. Adv. Energy Mater. 2025, 2405622
作者介绍
井立强,黑龙江大学教授,博士生导师。入选多项国家级人才计划、享受国务院政府特殊津贴专家、教育部创新团队带头人。现任黑龙江大学党委常委、副校长、功能无机材料化学教育部重点实验室副主任。现为中国可再生能源学会光化学专业委员会、中国感光学会光催化专业委员会和中国化工学会化工新材料专业委员会副主任委员等;担任“Materials Research Bulletin”、“The Innovation”、“Chinese Journal of Catalysis”、“Chinese Chemistry Letter”和“eScience”国际重要(SCI)刊物等编委。多年来主要围绕环境与能源光催化领域开展研究,主持承担20余项省部级以上重要课题,共获省科技奖一等奖2项、中国授权发明专利17项。作为通讯或第一作者,至今已在“Nat. Commun.”、“Angew. Chem. Int. Ed.”、“Adv. Mater.”、“Chem. Soc. Rev.”、“Energy Environ. Sci.”、“Adv. Energy Mater.”、“Adv. Sci.”和“Environ. Sci. Tech.”等上发表SCI论文200余篇,被SCI论文总引用18000余次,近10年连续成为了Elsevier公司发布的中国高被引学者。
张紫晴,黑龙江大学副教授,黑龙江省优秀青年基金获得者。2018年于吉林大学无机合成与制备化学国家重点实验室获得博士学位。主要从事有机聚合物基光催化剂的开发及其在光催化CO2还原和H2O2生产方面的研究工作。主持国自然青年项目在内的科研项目4项,作为第一作者及通讯作者至今在Nat. Commun.、Angew. Chem. Int. Edit.、Adv. Mater.、Appl. Catal. B等国际高水平期刊发表SCI论文22篇。
李卓,黑龙江大学讲师,黑龙江省优秀青年基金获得者。近年来主要从事有机半导体光催化相关工作。主持国自然青年项目在内的科研项目4项,以第一作者和通讯作者(包括共同)于Nat. Commun.、Angew. Chem. Int. Edit、Adv. Mater.、Adv. Energy Mater.、Adv. Sci.等著名国际期刊发表多篇研究论文。

