
全文速览
太阳能驱动的甲烷高选择性转化为高附加值化学品仍是一个长期存在的挑战。基于此,本文设计了紧密相连的原子级分散Fe物种和超小纳米Au负载的具有氧空位的SrTiO3中空纳米管(STOv),实现了CH4高选择性转化为CH3OH。最佳光催化剂Fe-Au/STOv实现了7.53 mmol g-1h-1的CH3OH产率,选择性高达95.4%。在365 nm波长下的表观量子效率达15.8%。实验和理论结果表明,SrTiO3表面构建的氧空位促进了CH4吸附,进而与光生空穴反应生成甲基自由基(•CH3)。同时光生电子可被锚定的Au迅速提取,然后转移到相邻的单原子Fe位点,活化O2生成关键中间体 Fe-*OOH,从而实现CH3OH的高选择性生成。本研究为设计高效光催化甲烷转化催化剂提供了新的位点协同调控策略和机理洞察。

背景介绍
甲烷作为重要的化工原料和强效温室气体,在温和条件下高选择性转化具有重要意义。目前工业上主要通过甲烷重整、费托合成的间接路线利用甲烷资源,其温度高达700 °C以上,其带来的高能耗不利于可持续发展。光催化技术具有巨大的潜力,可以克服反应势垒,为温和条件下甲烷直接转化为甲醇提供绿色途径。目前,已有一些关于CH4直接氧化为C1含氧产物的报道,其中宽带隙半导体(例如TiO2、ZnO、SrTiO3等)仍然是驱动此类反应的主要光催化剂。SrTiO3因其负导带和浅价带特性,可抑制H2O氧化生成游离且过量的•OH,为避免产物过氧化提供了机会。但其仍面临CH4吸附活化困难、光生电荷分离效率低及O2活化控制的三大挑战。因此,本研究通过纳米结构调控、构建表面氧空位和负载助催化剂,最终设计并合成了紧密连接的Fe-Au位点负载的氧空位SrTiO3中空纳米管(Fe-Au/STOv)催化剂,实现了光催化甲烷选择性转化为甲醇。
本文亮点
1) 发展了乙酰丙酮调控的静电纺丝策略并制备了形貌可控的一维SrTiO3中空纳米管,有利于电荷传输、CH4吸附和光捕获。进一步结合NaBH4介导的真空氢还原策略,以精确构建表面氧空位,同时抑制深层缺陷Ti3+的形成,进一步促进了电荷分离、CH4的吸附和活化。
2) 发展了一种逐步冷冻光还原诱导相邻金属位点定位沉积的策略,以精确地将紧密相连的Fe-Au物种锚定在氧缺陷 SrTiO3 中空纳米管上,从而有效地提取光生电子并通过调控 O2吸附构型,以稳定活性氧物种。
3) 最佳催化剂Fe-Au/STOv的CH3OH产率可达7.53 mmol g-1 h-1,选择性高达 95.4%,在365 nm波长处的表观量子产率(AQY)可达15.8%。
4) Fe-Au/STOv具有双重功能:(i)促进CH4的吸附以及由光生空穴介导的•CH3自由基的形成;(ii)实现O2的活化以生成关键的Fe-*OOH中间体。通过Au与Fe的协同作用实现了高达67.5%的优异的电子转移效率和较高的CH3OH选择性。
图文解析
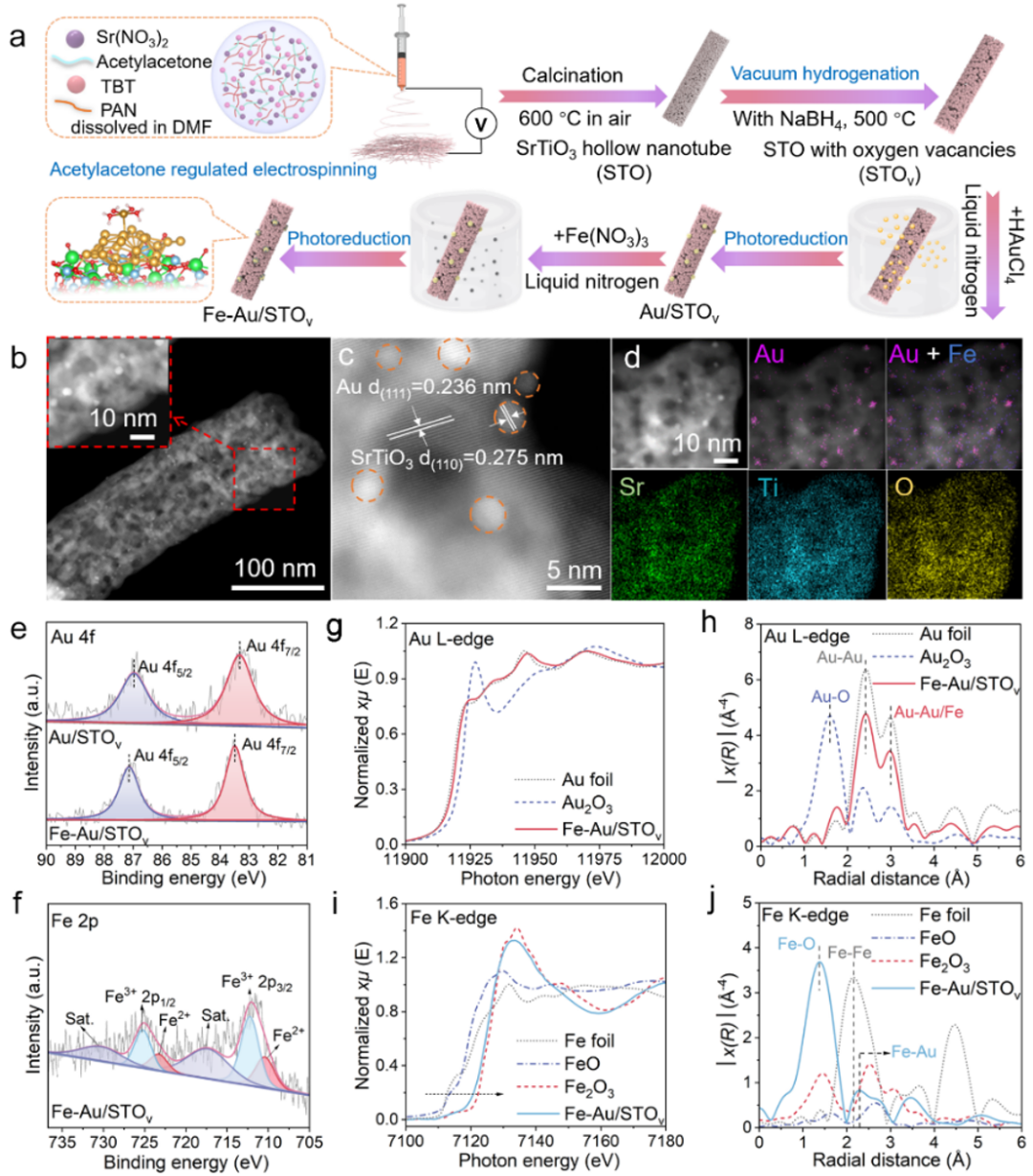
图1. Fe-Au/STOv的合成方法及结构表征。(a) Fe-Au/STOv设计和合成过程的示意图。(b-d) 具有不同标尺的Fe-Au/STOv的AC-HAADF-STEM图像以及Sr、Ti、O、Au和Fe元素的EDX mapping 图像。(e) Au/STOv和Fe-Au/STOv的Au 4f XPS分析。(f) Fe-Au/STOv的Fe 2p XPS分析。(g, h) Fe-Au/STOv上Au L3-edge的XANES和EXAFS谱。(i, j) Fe-Au/STOv上Fe K-edge的XANES和EXAFS谱。
球差电镜证明超小的Au纳米颗粒(2-3 nm)均匀分布在一维STOv中空纳米管表面,XPS分析表明引入的Au与STOv具有相互作用。XPS和XANES共同证明Au主要以金属态存在,Fe物种的化学态以+3价为主。EDX mapping、XPS和Au与Fe的EXAFS结果综合表明Fe单原子主要锚定在Au纳米颗粒上。
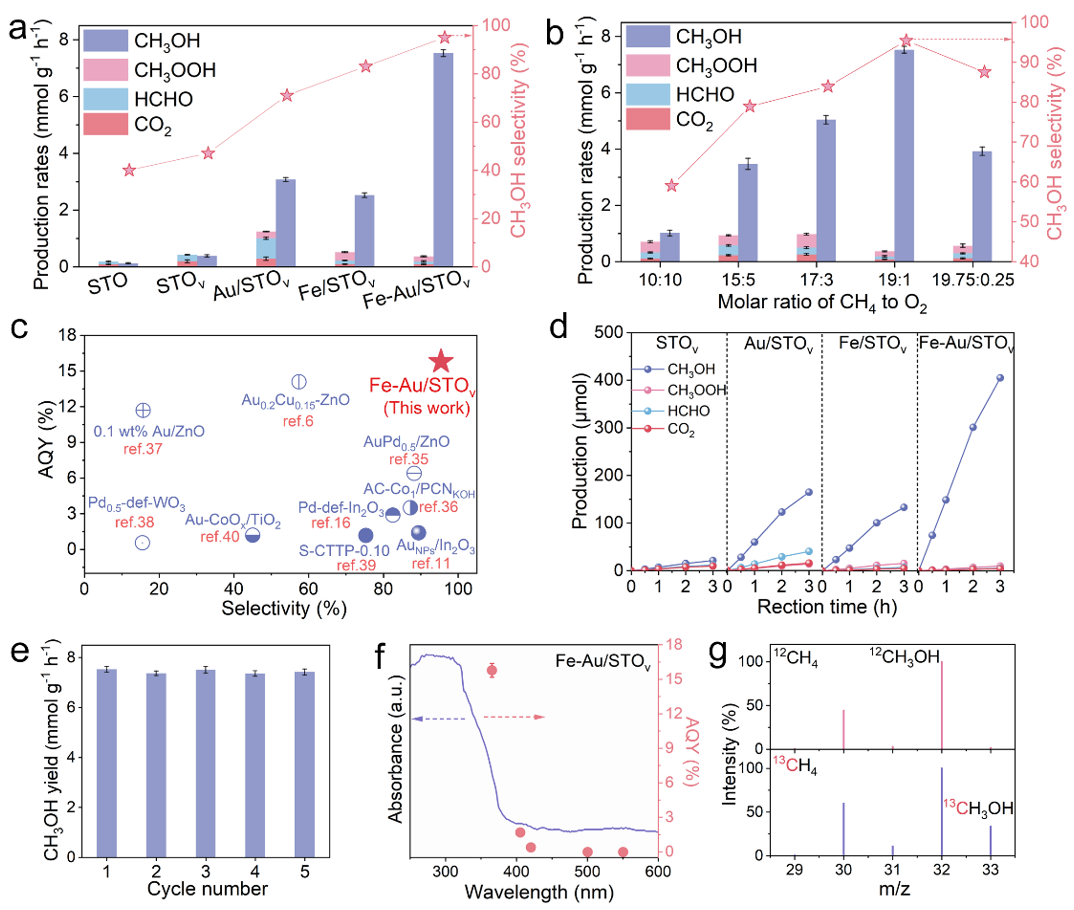
图2. 光催化甲烷转化的活性和选择性。(a) STO、STOv、Au/STOv、Fe/STOv和Fe-Au/STOv光催化CH4转化的性能。反应条件:20 mg催化剂,30 mL H2O,CH4/O2 = 19:1 (bar),25 °C。(b) 不同CH4与O2摩尔比下Fe-Au/STOv的甲醇产率和选择性。(c) Fe-Au/STOv与已报道的光催化剂的CH3OH产率和选择性比较。(d) STOv、Au/STOv、Fe/STOv和Fe-Au/STOv上CH3OH、CH3OOH、HCHO和CO2随时间变化的产率。(e) Fe-Au/STOv的循环稳定性。(f) Fe-Au/STOv上CH3OH生成的AQY随入射光波长的变化。(g)同位素标记的质谱。
使用O2作为氧化剂评价了所研究样品的光催化CH4转化性能。优化后得到的催化剂Fe-Au/STOv具有最佳的光催化CH4选择性氧化性能,实现了7.53 mmol g-1h-1的CH3OH产率和高达95.4%的选择性。同时Fe-Au/STOv展现出良好的循环稳定性。在365 nm单波下,Fe-Au/STOv的表观量子效率(AQY)达到了15.8%,与已报道的可比条件下的光催化剂相比,具有显著优势
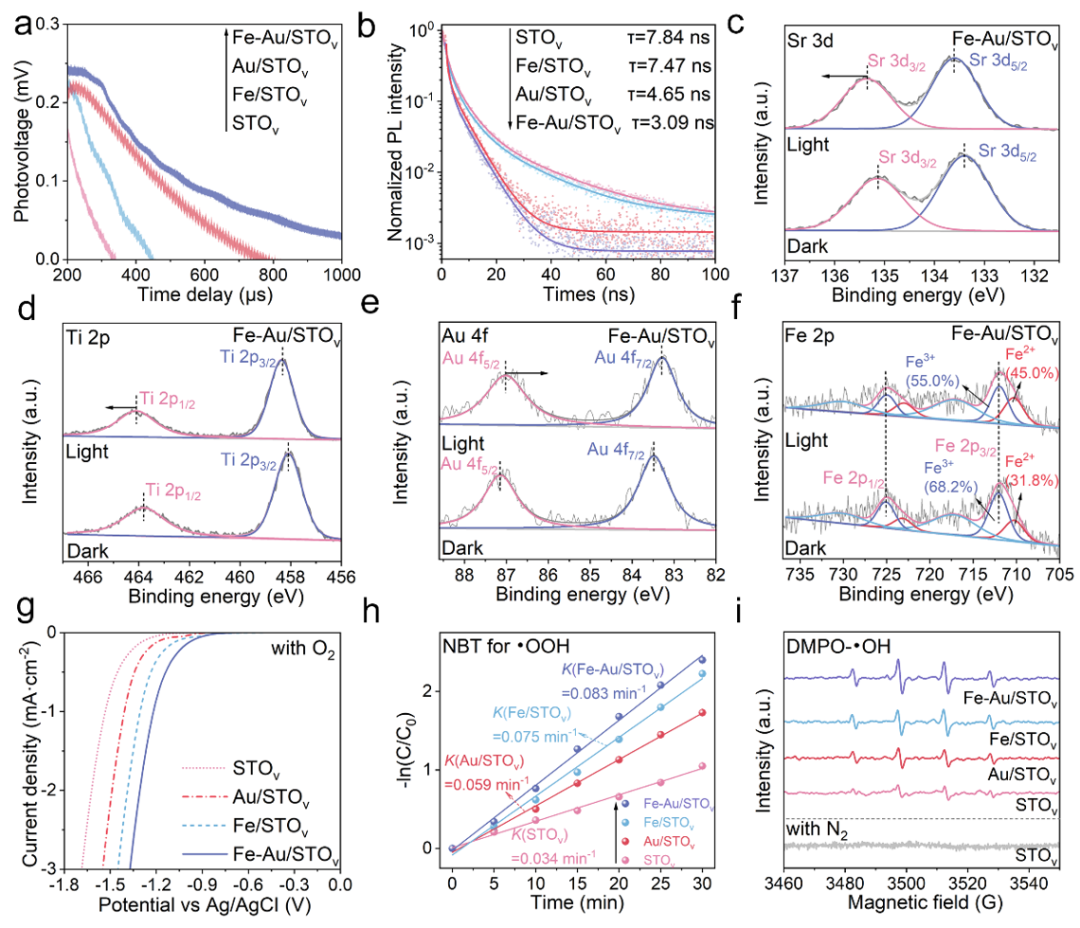
图3. 电荷分离和转移以及涉及的ROS机制研究。 (a) STOv、Fe/STOv、Au-STOv和Fe-Au/STOv的TPV响应和(b) 时间分辨PL谱。(c-f) Fe-Au/STOv在黑暗和光照下Sr 3d (c)、Ti 2p (d)、Au 4f (e) 和Fe 2p (f) 的原位光照XPS分析。(g) STOv、Fe/STOv、Au/STOv和Fe-Au/STOv在O2饱和电解质中的电化学还原曲线。(h) 不同光催化剂上用于检测·OOH自由基的NBT光降解动力学常数。(i) EPR谱图。
瞬态光致发光(TR-PL)光谱、瞬态表面光电压谱(TPV)测试表明Au和Fe可以有效地促进STOv的电荷分离。进一步利用原位光照XPS研究Fe-Au/STOv的电荷转移方向。相较于暗态,光照后Sr和Ti的结合能均变大,而Au 4f的结合能偏小,说明Au可接受来自STOv的光生电子。类似地,Fe 2p在光照后Fe3+的峰含量降低,说明Fe物种在光照过程中进一步接收Au转移的光生电子,导致Fe3+含量降低,Fe2+含量升高。通过O2饱和电解质中的电化学还原曲线,NBT监测•OOH自由基和EPR捕获•OH自由基证明,Fe-Au物种可以有效促进O2的还原生成ROS(•OOH和•OH)。以上结果共同证明了Au和Fe物种在电荷分离与转移和O2活化生成ROS方面具有协同促进作用。

图4. 通过原位TAS研究电子动力学图。 (a) STOv、Fe/STOv、Au/STOv和Fe-Au/STOv在Ar (H2O)中的原位μs-TAS谱图。(b)不同样品中寿命变化和ETE值的比较。(c-e) 在CH4 (H2O) 和 CH4 (O2 + H2O) 条件下,Au/STOv (c)、Fe/STOv (d) 和 Fe-Au/STOv (e) 的μs-TAS衰减动力学。(f)所研究样品的半衰期(t50%)、ETE、AQY、选择性和CH3OH产率的关系图。
利用原位瞬态吸收光谱揭示了负载的Fe-Au物种可有效地捕获STOv产生的光生电子,促进CH4氧化反应中的O2还原动力学过程。在Ar (H2O)条件下,通过半衰期(t50%)的寿命对比说明Au与Fe物种相比具有更强的电子捕获能力,并且Au和Fe的共修饰对STOv的电子转移具有协同促进作用。因此,通过与Ar(H2O)条件对比,研究了O2还原过程的电子转移效率(ETE),证明了Au和Fe物种可以协同增强电子转移,从而促进O2活化。此外,进一步研究了CH4氧化过程中O2还原的电子转移动力学。最佳光催化剂Fe-Au/STOv在模拟CH4氧化的原位反应条件下电子转移效率可达67.5%,远高于Au/STOv (13.6%)和Fe/STOv (27.9%)。

图5. 光催化CH4选择性氧化为CH3OH的机理及DFT。(a) STO和STOv在暗态条件下对CH4和H2O吸附的原位红外光谱。(b) STOv、Fe/STOv、Au/STOv和Fe-Au/STOv的O2程序升温脱附曲线。(c) Fe-Au/STOv在暗态条件下对O2吸附的原位红外光谱。(d) Fe-Au/STOv的CH4氧化过程中检测到的中间体。(e) Fe-Au/STOv的原位拉曼光谱。(f) Au/STOv和Fe-Au/STOv对O2吸附和还原的相关自由能。(g) CH4氧化制CH3OH反应路径的自由能图以及CH3OH进一步氧化与解吸竞争的能垒图。
原位红外和理论计算结果揭示了STO表面的氧空位可以促进CH4吸附。通过O2-TPD证明Fe-Au物种的负载可以促进O2的吸附。进一步通过原位红外、原位拉曼和理论计算结果证明Fe单原子修饰后改变了O2在STOv上的吸附位点和吸附构型。O2在Fe-Au位点上形成 “end-on”吸附构型,被光生电子还原形成吸附态的Fe-*OOH中间体,从而促进 CH3OH生成同时避免过氧化。

图6. CH4在Fe-Au/STOv催化剂上光催化选择性氧化为CH3OH的示意图。
SrTiO3表面构建的氧空位可以促进CH4吸附,从而有效捕获光生空穴产生•CH3,与此同时,光生电子会被修饰的Au快速提取,并迅速转移至相邻的单原子Fe位点进而活化O2。O2分子以“end-on”构型吸附在Fe位点上发生转化,形成关键的Fe-*OOH中间体。重要的是,通过电子还原O2产生温和可控的ROS(•OOH)自由基及其在Fe位点上的稳定,有效抑制了产物的过氧化,从而对高选择性生成CH3OH具有关键作用。
总结与展望
综上,本研究发展了原子级分散Fe物种和超小Au纳米颗粒(2-3 nm)共负载的STOv催化剂,应用于光催化甲烷选择性氧化制甲醇。最佳催化剂Fe-Au/STOv实现了7.53 mmol g-1h-1的甲醇产率和95.4%的选择性。系列原位实验和DFT计算表明:SrTiO3表面构建的氧空位促进了甲烷吸附,从而有效捕获光生空穴产生甲基自由基;而光生电子可被锚定的Au快速提取并转移至相邻Fe位点高效活化O2,得益于O2的“end-on”吸附构型形成的稳定Fe-*OOH中间体,有效抑制了产物过氧化。该研究揭示了甲烷氧化中CH4与O2协同活化机制,为甲烷可持续光催化转化领域的活性位点精准设计和电子动力学研究提供了新途径。

