石墨氮化碳(g-C3N4)在H2O2的人工光合成中获得了越来越多的关注,但其性能却受到氧还原反应(ORR)动力学缓慢和电子寿命短的阻碍。
2024年10月25日,黑龙江大学副校长井立强教授、边辑副研究员、张紫晴副教授团队在Nature Communications期刊发表题为“Photocatalytic H2O2 production over boron-doped g-C3N4 containing coordinatively unsaturated FeOOH sites and CoOx clusters”的研究论文,团队成员Liu Ping、Liang Teng为论文共同第一作者,张紫晴副教授、边辑副研究员、井立强教授为论文共同通讯作者。
该研究展示了一种用配位不饱和FeOOH和CoOx簇定制的B掺杂g-C3N4(BCN),用于在没有牺牲剂的情况下从水和氧中进行H2O2光合成。与g-C3N4相比,最佳材料在可见光下的活性提高了30倍,太阳能到化学物质转化效率达到0.75%,在已报道的g-C3N4基光催化剂中名列前茅。此外,原位微秒瞬态吸收光谱显示,氧还原反应的电子转移效率达到34.1%。实验和理论结果表明,CoOx引发空穴-水氧化并延长电子寿命,而FeOOH则接受电子并促进氧活化。有趣的是,直接一步双电子反应生成H2O2的关键在于配位不饱和FeOOH,它可以调节O2的pauling型吸附构型,以稳定过氧化物,抑制超氧自由基的形成。

该研究报道了一类催化剂(CoOx-BCN-FeOOH),用于可见光下在水和氧混合物中进行H2O2光合成,无需任何牺牲剂。与g-C3N4相比,最佳方案的活性提高了30倍,SCC效率高达0.75%。研究发现CoOx簇有利于空穴-水氧化,延长了BCN的电子寿命。而FeOOH被证明可以接受电子并促进氧的活化。此外,通过实验和理论方法阐明了配位不饱和Fe位点调控的一步双电子ORR途径。该研究为开发具有双活性位点工程的高效光催化剂生产H2O2提供了设计指导,并为研究ORR过程中的电子动力学提供了方法。
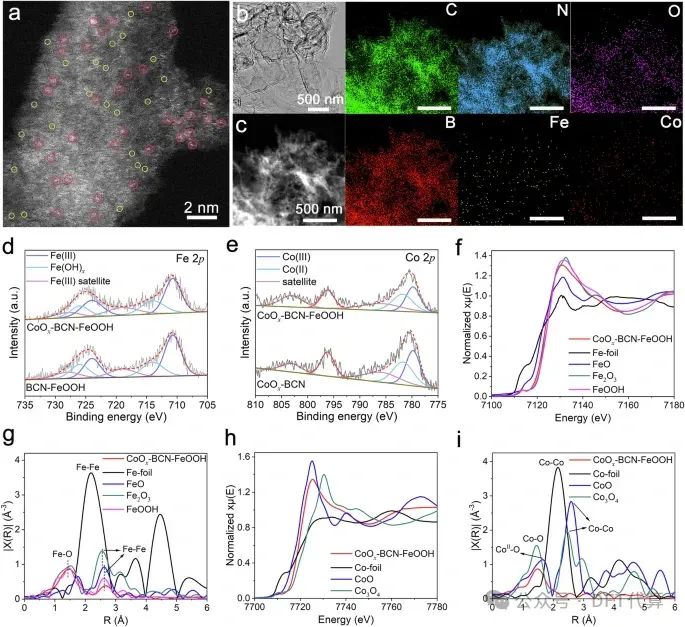
图1. CoOx-BCN-FeOOH的形貌和结构表征。a像差校正高角环形暗场扫描TEM(AC-HAADFSTEM)图像,b CoOx-BCN-FeOOH的TEM图像,c HAADF-STEM图像以及相应的c、N、O、b、Fe和Co的EDX映射图像(比尺:500 nm)。d BCN-FeOOH和CoOx-BCN-FeOOH中Fe 2p XPS分析。e CoOx-BCN和CoOx-BCN-FeOOH的Co 2p XPS分析。f Fe箔、FeO、Fe2O3、FeOOH和CoOx-BCN-FeOOH的Fe K边XANES谱图。g CoOx-BCN-FeOOH的Fe k边EXAFS谱拟合曲线。h Co箔、CoO、Co3O4和CoOx-BCN-FeOOH的Co k边XANES谱图。i CoOx-BCNFeOOH Co k边EXAFS谱拟合曲线。
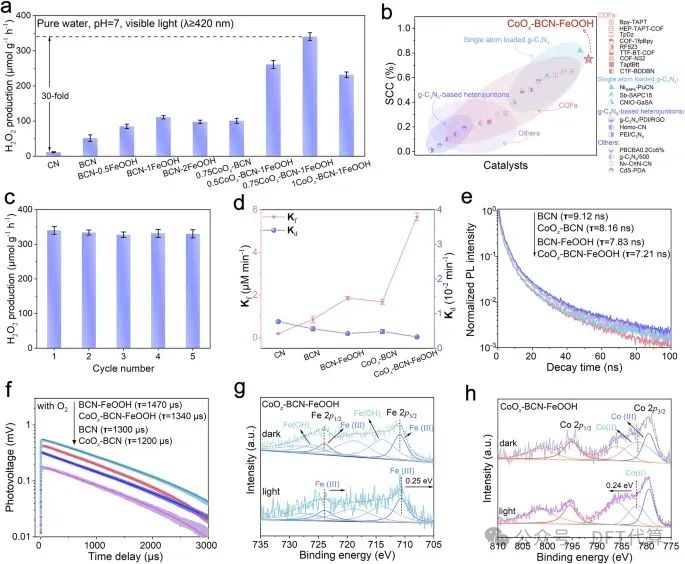
图2. H2O2光合成性能及CoOx和FeOOH在电荷调制中的作用。a在可见光照射下(pH=7.0),CN、BCN、BCN-FeOOH、CoOx-BCN和CoOx-BCN-FeOOH对纯水制取H2O2的光催化性能。b CoOxBCN-FeOOH与最近报道的光催化剂的SCC效率比较。c可见光照射下,CoOx-BCN-FeOOH用纯水连续5次制H2O2。d CN、BCN、BCN-FeOOH、CoOx-BCN和CoOx-BCN--FeOOH的H2O2生成速率常数Kf和分解速率常数Kd。e在O2气氛中BCN、BCN--FeOOH、CoOx-BCN和CoOxBCN-FeOOH的时间分辨PL光谱和f TPV响应。g-h CoOx-BCN-FeOOH中Fe 2p和Co 2p的原位辐照XPS分析。
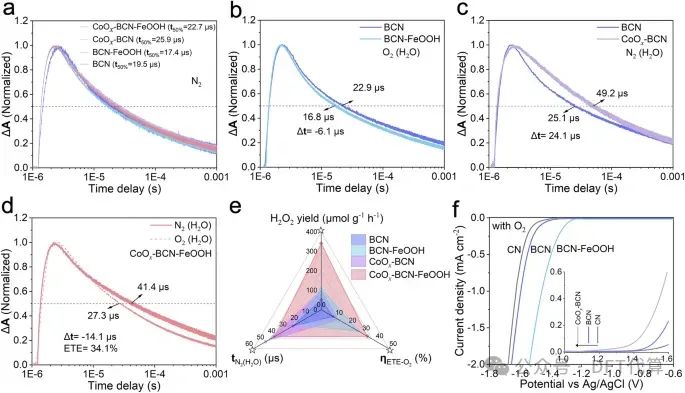
图3. 原位TAS对ORR电子动力学的研究。a BCN、BCN-FeOOH、CoOx-BCN和CoOx-BCN-FeOOH在N2中的μs-TAS衰变动力学。b BCN和BCN-FeOOH在O2(H2O)条件下的μsTAS衰变动力学。BCN和CoOx-BCN在N2(H2O)条件下的c μs-TAS衰变动力学。CoOx-BCN-FeOOH在不同气氛下的d μs-TAS衰变动力学。e所研究催化剂的半衰期(t50%)、电子转移效率(ETE)与H2O2产率的关系图。f在O2饱和电解质中CN、BCN和BCN-FeOOH的电化学还原曲线(CN、BCN和CoOxBCN的氧化曲线见插图)。
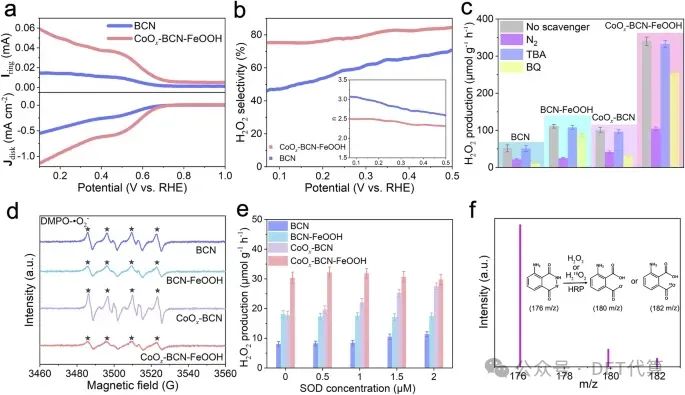
图4 . H2O2光合成的一步两电子ORR途径。a在饱和的0.1 M KOH溶液中,在1600 rpm下,在环形电流(上)和圆盘电流(下)下,BCN和CoOx-BCN-FeOOH的RRDE极化曲线。b H2O2选择性作为应用电位的函数(插图显示了计算的平均转移电子数)。c一系列样品在不同反应气体或不同牺牲剂作用下的H2O2生成速率。d光照射下BCN、BCN-FeOOH、CoOx-BCN和CoOx-BCN-FeOOH的DMPO自旋俘获EPR谱。e不同超氧化物歧化酶(SOD)浓度下BCN、BCN-FeOOH、CoOx-BCN和CoOx-BCN-FeOOH的H2O2生成速率。f在CoOx-BCNFeOOH上用H218O光催化生成的过氧化氢氧化后鲁米诺的质谱。
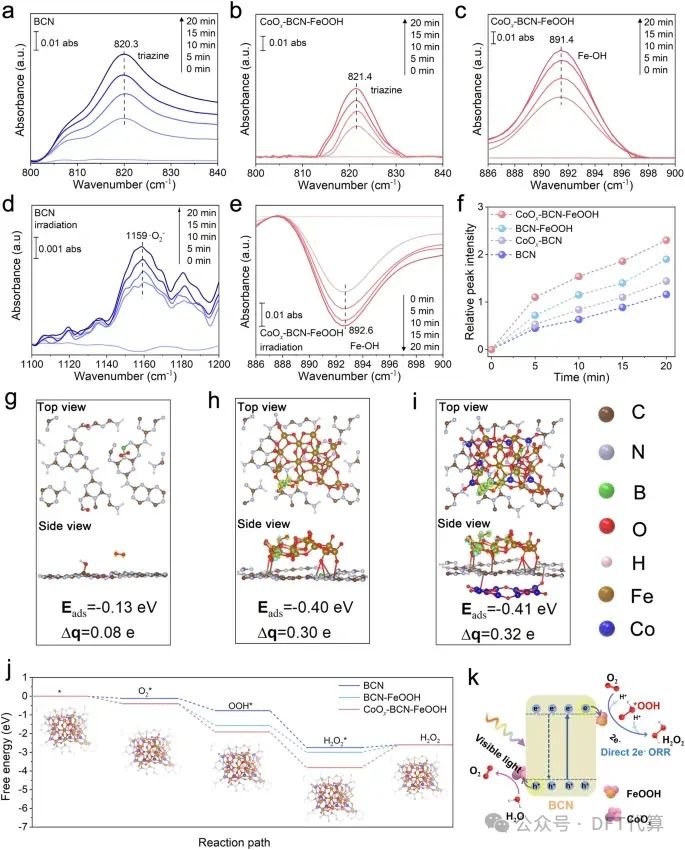
图5. H2O2在CoOx-BCN-FeOOH上的光合成机理。a-c BCN和CoOx-BCN-FeOOH对O2吸附的原位DRIFTS。d-e光照射下BCN和CoOx-BCN-FeOOH的原位DRIFTS。f不同样品产生H2O2的相对峰值强度。g-i BCN、BCN-FeOOH和CoOx-BCNFeOOH上O2吸附的不同电荷密度构型俯视图和侧视图。Eads和Δq分别为O2的总吸附能和O2上的电荷转移。黄色表示电子积累,浅蓝色表示耗尽。j BCN、BCN-FeOOH和CoOx-BCN-FeOOH上2e-ORR过程的自由能图以及每步优化构型,星号(*)表示催化剂的活性位点。k CoOx-BCN-FeOOH的电荷转移和氧化还原反应示意图。
总之,该研究提出了一种直接一步2e-ORR的策略,可以在没有任何牺牲剂的情况下有效地生产H2O2。配位不饱和FeOOH和CoOx簇被精确地锚定在BCN上,优化后的CoOx-BCN-FeOOH光催化剂在可见光照射下产生H2O2的速率为0.34 mmol g−1 h−1,SCC效率为0.75%。原位TAS方法表明,CoOx-BCN-FeOOH的ORR电子转移效率为34.1%。实验和理论结果证实,该催化剂产生H2O2的优异活性是由于引入的FeOOH的氧活化和改性的CoOx簇在延长电子寿命方面的协同作用。重要的是,FeOOH的配位不饱和结构允许O2适应pauling型吸附构型,进一步稳定过氧化物,限制超氧自由基的形成,这有助于直接一步2e−ORR途径的高选择性。该研究提供了一种双活性位点工程策略,以调节单组分催化剂的电子寿命和氧活化,从而高效生产H2O2。

